Изобретение относится к химии производных арилсульфонилмочевины, в частности, к усовершенствованию способа синтеза соединений общей формулы
ArSO2NHCONHR',
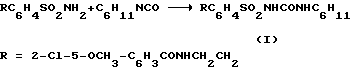
где R = ArCONHCH2CH2, HtCONHCH2CH2, R' = циклоалкил, циклоалкенил, в том числе замещенные циклоалкил- или циклоалкенил.
К соединениям этого класса относится большая серия дизамещенных сульфонилмочевин, заявленных как противодиабетические средства [Neth, Appl. 6.610.580 (1965), 6.603.398 (1966); Ger.Offen DE 1.185.180 (1965), 2.012.138 (1970); US 3.932.503 (1976); US 3.917.690 1975; Fr.Dem. 2.138.112, 1973; JP 4.9051.286 (1974)/.
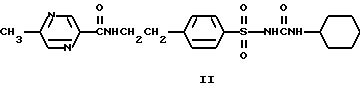
Среди них 1-{4-[2-(2-метокси-5-хлорбензамидо)этил]фенилсульфонил}- 3-циклогексилмочевина (глибурид, глибенкламид, манинил) (I) и 1-{4-[2-(5-метилпиразин-2-карбоксамидо)этил] фенилсульфонил} -3- циклогексилмочевина (глипизид, минидиаб, глибинез) (II)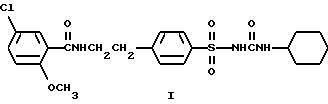
противодиабетические препараты, инсулиновые секретогены второго поколения сульфонилмочевин, современные средства для лечения сахарного диабета. Машковский М.Д. Лекарственные средства. Изд. 13, Харьков; Торсинг, 1998, т. 2, с. 23. Черных В.П. Хим. фарм. Журнал 1978, т. 12, N 8, с. 13-20. Almuer W. et. al. Arzneim. Forsch. 1966, BD 16, N 12. С. 1640-1642. Challinor-Rogers J.L. et. al. J.Pharmacol. Exp. Ther. 1955, v. 273, N 2, p. 778-786.
Синтез обычно выполнялся путем преобразований концевой функциональной группы в соответствующем сульфонамиде, в частности, в случае глибурида в N-[2-(4-аминосульфонил)фенилэтил]-2-метокси-5- хлорбензамиде (III), глипизида - в N-[2-(4-аминосульфонил)фенилэтил] - 5-метилпиразин-2-карбоксамиде (IV). Эти способы многочисленны, но чаще используют уретановый и изоцианатный варианты. Именно по этим методикам синтезирована огромная серия сульфонилмочевин, в том числе близких аналогов глибурида и глипизида.
В уретановом способе синтеза чаще всего применяют первоначальный перевод сульфонамида типа (III, IV) в соответствующую сульфонилмочевину "A", далее сульфонилмочевина может быть введена в реакцию с метанолом с образованием уретана "B":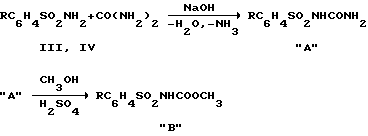
который выделяется, а затем вводится в конденсацию с амином, например, как в случае соединений (I, II), с циклогексиламином (например, кипячение уретана с циклогексиламином в ксилоле) Challinor-Rogers J.L. et. al. J. Pharmacol. Exp. Ther. 1995, Vol 273, N 2, p. 778-786.
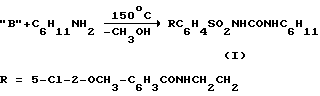
Недостатки способа: он многостадиен, требует больших энергозатрат, суммарный выход препаратов на завершающей стадии не превышает 70%, при этом требуется применение достаточно большого ряда реагентов.
Более прост и экономичен изоцианатный вариант, базирующийся на реакции сульфонамида типа (III) с изоцианатом в присутствии оснований [ Neth Appl. 6610580 C 07 C, 1965 [C.A. 1968, Vol. 68, 12 733h. DD 204915 C 97 C 143/833, 1983. Описан способ синтеза глибурида при нагревании в смеси ацетона с ДМФА при применении поташа [DD. 20 4915 C 07 C 143/833, 1983].
Согласно патенту [6] , 41 г (0.11 моль) сульфонамида (III) и 0.22 моль поташа в смеси 80 мл ДМФА и 220 мл ацетона обрабатывают 15.5 мл (0.12 моль) циклогексилизоцианата (VI) в 20 мл ацетона и кипятят 5 ч. Калиевую соль отделяют, промывают 50 мл ацетона, кипятят 30 мин в 500 мл метанола (этанола), охлаждают до 30oC, фильтруют, промывают 50 мл спирта, переносят в 800 мл воды, подкисляют суспензию разбавленной соляной кислотой до pH1, выдерживают при комнатной температуре 3 ч, отфильтровывают, промывают водой до нейтральной реакции, сушат и получают 52.5 г технического продукта с т.пл. 171-172oC, содержащего 0.2% исходного сульфонамида. Продукт очищен при кипячении в смеси ДМФА-ацетон (360 и 66 мл соответственно) с активированным углем с образованием глибурида (46 г), содержащего лишь следы исходного вещества, или кристаллизацией из дихлорэтана. Суммарный выход - 80 и 83%, соответственно, т.пл. 173-174oC.
Недостатки способа.
Метод не технологичен:
1) на стадии синтеза и очистки применяется смесевой растворитель (ацетон-ДМФА), что делает практически невозможной его регенерацию, при этом расход растворителей значителен (19.4 об.ч. смесевого растворителя и 13.4 спирта на 1 в.ч. амида (III));
2) метод длителен и энергоемок: реакция идет гетерогенно: поташ плохо растворим в ДМФА и очень плохо - в его смеси с ацетоном даже при температуре кипения; скорее всего так же плохо растворима в этой смеси калиевая соль исходного сульфонамида, образующегося на первом этапе реакции; гетерогенность процесса предопределяет его длительность (5 ч) и необходимость нагревания;
3) по мере образования калиевой соли сульфонамида (III) и при последующем ее переходе в калиевую соль целевого вещества (I) вся масса принимает вязкую сметаноподобную консистенцию, плохо контролируется, трудно перемешивается, практически не льется и плохо фильтруется; при переносе соли в метанол образуется также пастообразная масса с теми же особенностями, хотя модуль по метанолу достаточно высок (1:10); при переносе отфильтрованного вещества в воду вновь образуется густая суспензия, т.е. последующее подкисление с переводом образовавшейся соли в глибурид выполняется гетерогенно (кристаллическая соль переводится в кристаллический же глибурид) и плохо поддается контролю;
4) по ходу выполнения реакции требуются фильтрации вязкой высокодисперсной суспензии, что неизбежно ведет к значительным механическим потерям целевого продукта.
Наиболее близким аналогом предлагаемого нами решения является более технологичный вариант изоцианатного синтеза сульфонилмочевин, в частности, глибурида, из соответствующих арилсульфонамидов в смеси ацетона и водной щелочи при 0-5oC [DD. 204915, C 07 C 143/833].
В этом варианте к 9.1 г (0.025 моль) арилсульфонамида (III) в 12.5 мл 2 н. раствора NaOH (0.025 моль в виде 8%-ного раствора) и 30 мл ацетона при 0-5oC добавляют по каплям 3.3 г (0.0264 моль) циклогексилизоцианата. Через 3 ч смесь разбавляют водой и метанолом, фильтруют и подкисляют соляной кислотой.
Процесс включает единственную целевую стадию и, на первый взгляд, достаточно прост.
Однако следует отметить, что:
1) конверсия сульфонамидов типа (III) в целевые вещества, по нашим данным, как и выход технических продуктов по этим методикам составляет 60-70%, и уже только из этих фактов a priori следует ожидать значительную примесь (более 20%) непрореагировавшего амида в продукте реакции; это и подтверждается в эксперименте: выход сульфонилмочевин, обычно ниже заявленного, препараты очень загрязнены примесями, в них в большой доле присутствует исходный амид, вещества требуют дополнительной и часто неоднократной очистки;
2) в качестве примеси в целевых веществах помимо исходного амида присутствует еще один продукт - соответствующая диалкилмочевина, при синтезе соединений (I, II) - эти дициклогексилмочевина (V); количество этой примеси достигает в отдельных опытах 5-10%.
По ходу синтеза в описанных условиях наблюдается газовыделение и это может объясняться тем, что изоцианаты в водном ацетоне наряду с основной реакцией подтверждены побочной: присоединяют воду с образованием карбамата натрия, который может быть источником карбаминовой кислоты; последняя нестабильна и распадается с образованием углекислоты и, главное, соответствующего амина "C" [в случае синтеза мочевин (I, II) - циклогексиламина];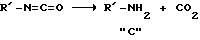
где R' - циклоалкил, циклоалкенил, в том числе замещенные циклоалкил- или циклоалкенил. Образование амина ("C") предопределяет его взаимодействие с исходным изоцианатом с образованием диалкил(дициклогексил)мочевины, в случае синтеза соединений (I, II) это: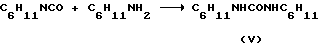
Таким образом, и образование в этом синтезе побочных продуктов - диалкилмочевин типа (V), и присутствие заметных количеств непрореагировавших сульфонамидов обусловлено заметным разложением одного из реагентов - изоцианата, в водной среде (водный ацетон), т.е. самим способом реализации этой реакции. Основная реакция, в отличие от варианта [6], идет быстро, так как выполняется в гомогенной среде (натриевые соли сульфонамидов растворимы лишь в водных растворителях). Отметим, что попытки более полно использовать в реакции сульфонамиды типа (III) при введении в синтез избытка изоцианата приводит лишь к повышению доли диалкилмочевин типа (V) в ее продуктах.
То, что вещество (I), полученное по этому способу, не отвечает требованиям по качеству свидетельствует целая серия самостоятельных патентов, посвященных его очистке (например, CA 889876, 1972, DD 248115).
Основные способы очистки - превращение технического глибурида в соли и последующее его выделение при подкислении. Например, продукт обрабатывают метилатом натрия и кристаллизуют соль из уксусной кислоты или продукт превращают в соль амина в органическом растворителе, фильтруют и обрабатывают полученный раствор кислотой. Последний вариант для глибенкламида, содержащего 8% дициклогексилмочевины и 0.4% исходного сульфонамида, включает растворение в метаноле, дозировку бутиламина при охлаждении, фильтрацию раствора и обработку его уксусной кислотой (выход - 86%).
Во всех случаях очистки используются дополнительные реагенты, часто весьма дорогостоящие, при этом потери продукта всегда значительны (15-20%).
Отметим, что исходные сульфонамиды, побочные диалкилмочевины и целевые сульфонилмочевины относятся к NH-кислотам, которые обладают достаточно близкой кислотностью; все они в приведенных условиях дают соли, генерирующие при подкислении те же вещества.
Таким образом, при солевых способах очистки заведомо нет никакой гарантии четкого разделения продуктов, особенно в тех случаях, когда доля примесей значительна. Они могут применяться с успехом лишь тогда, когда количество примесей незначительно, т.е. сам способ синтеза должен обеспечивать получение достаточно чистого технического препарата.
Суммируя изложенное, следует отметить, что метод наряду с простотой и технологичностью обладает и существенными изъянами.
Недостатки способа-прототипа:
1) недостаточно высокий выход технического продукта (не выше 70-75%);
2) препараты включают примеси, присутствие которых предопределено применяемой технологией;
3) по качеству препараты, например, глибурид и глипизид, существенно уступают фармакопейным из-за присутствия в технических продуктах значительного количества примесей, из-за чего целевые продукты требуют дополнительной очистки;
4) очистка технических препаратов в силу близости структуры и физико-химических свойств целевых и побочных продуктов затруднена, иногда она проводится неоднократно и всегда сопровождается значительными потерями.
Т.о., хотя применяемый в прототипе вариант синтеза весьма прост технологически, он не дает технические продукты достаточной степени чистоты и с высоким выходом.
Из анализа всех приведенных данных следует, что для получения кондиционного продукта по изоцианатному способу при применении достаточно простой технологии должны обеспечиваться условия, в которых не только исходный сульфонамид типа (III), но и изоцианат максимально расходуется по целевому направлению и дициклогексилмочевина (V) не образуется. Этого можно достичь, если процесс выполняется быстро (в растворе) и гидролиз изоцианата максимально предотвращен (в отсутствие воды).
Задачей предлагаемого изобретения является создание на базе изоцианатного варианта синтеза технологически приемлемого процесса, обеспечивающего высокую чистоту технических сульфонилмочевин, в том числе глибурида и глипизида, и их высокий выход.
Задача может быть решена при разработке метода, обеспечивающего присоединение арилсульфонамидов типа (III, IV) к изоцианатам в условиях, сводящих к минимуму побочное разложение последнего водой с образованием соответствующего амина (циклогексиламина). Эти условия позволят практически исключить синтез побочного продукта - диалкилмочевины [дициклогексилмочевины (V)], обеспечат наиболее полное превращение исходных продуктов - изоцианатов и арилсульфонамидов, в целевые вещества и, как следствие, - достаточно высокий выход и качество технической сульфонилмочевин, в частности, глибурида и глипизида (I, II), не нуждающихся в сложной дополнительной очистке.
Сущность решения состоит в том, что стадию присоединения арилсульфонамидов к изоцианатам предлагается проводить при взаимодействии последних с органическими солями арилсульфонамидов в среде органических апротонных растворителей (ацетон, ацетонитрил, ДМФА, хлористый метилен, хлороформ, дихлорэтан и т.д.).
В качестве солей предлагается использовать высоколипофильные четвертичные аммониевые соли сульфонамидов, например, тетрабутиламмониевые, триалкил(C8-C10)метиламмониевые /Адоген 464/, метилтриоктиламмониевые /Аликват 336/, бензилтриэтиламмониевые и т.п., что обеспечивает полную растворимость соответствующих солей арилсульфонамидов в органической среде и позволяет работать в однофазной системе.
Реакцию предлагается проводить при комнатной температуре. Целесообразно использование не смешивающегося с водой растворителя, что упрощает обработку реакционной массы на стадии выделения целевого вещества: после промывки ее водой и разбавленной минеральной кислотой можно отогнать растворитель и вернуть его в цикл, а остаток обработать спиртом (метанол, этанол, 2-пропанол) и выделять продукт обычным способом; объединенный и перегнанный маточный и промывной спирт, полученный после отделения сульфонилмочевины на завершающей операции, можно использовать в обороте; эти приемы обеспечивают практически безотходный вариант технологического процесса.
В результате предлагаемого решения при проведении синтеза с использованием сульфонамида в виде его четвертичной аммониевой соли процесс реализуется по схеме: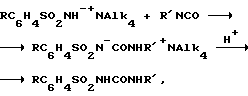
где Alk, например, CH3(CH2)3; R', например, циклогексил; R, например: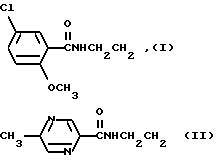
В традиционном варианте [5] используются натриевые соли сульфонамидов и водноацетоновая среда (для растворения соли сульфонамида); применяется охлаждение (0-5oC) /во избежание быстрого гидролиза изоцианата/.
В отличие от него при применении органических солей арилсульфонамидов ввиду их хорошей растворимости в органической среде становится возможным проведение синтеза в органическом апротонном растворителе в отсутствие воды. Это практически исключает гидролиз изоцианата, а следовательно - образование побочного продукта - диалкилмочевины, и снимает необходимость в охлаждении; проведение синтеза при комнатной температуре способствует более быстрому превращению сульфонамида в сульфонилмочевину, при этом технология упрощается, а энергозатраты снижаются.
Возможность проведения синтеза в растворителе, не смешивающемся с водой, облегчает последующую стадию промывки и выделения целевого продукта, и позволяет регенерировать растворитель полностью.
Именно эти условия обеспечивают -
1) быструю и высокую конверсию сульфонамида в сульфонилмочевину, а, следовательно, практическое отсутствие исходного сульфонамида в продуктах реакции;
2) высокую степень превращения изоцианата в целевую сульфонилмочевину, практическое отсутствие его гидролиза в соответствующий амин, т.е. малую вероятность их взаимодействия с образованием диалкилмочевины и, следовательно, ее отсутствие в продукте;
3) как следствие п.п. 1, 2 - достаточно высокий выход и чистоту технических продуктов (выход 85-95%, содержание основного вещества более 98%);
4) простоту и технологичность процесса и возможность максимальной утилизации отходов.
То, что образующаяся в виде органической соли сульфонилмочевина остается в органическом слое, удобно для последующей обработки: промывки водой и раствором кислоты.
Масса после удаления растворителя и обработки спиртом представляет собой легко подвижную суспензию, хорошо фильтруется, технические целевые продукты не имеют окраски и отвечают требованиям Британской и Европейской Фармакопей.
Для лучшего понимания сущности предполагаемого изобретения приводится примеры его конкретной реализации.
Общая методика синтеза:
К раствору 0.01 моль тетралкиламмониевой соли арилсульфонамида, например, тетрабутилаамониевой соли амидов (III, IV), в 50-60 мл хлористого метилена (хлороформа, дихлорэтана, ацетона, ацетонитрила, ДМФФ и т.п.) приливают 1.5 мл (0.012 моль) изоцианата (циклогексилизоцианата), выдерживают 15 мин и проверяют полноту превращения исходного сульфонамида (ТСХ); в случае необходимости выдержку увеличивают до 30 мин.
а. При работе в не смешивающемся с водой растворителе реакционную массу промывают водой (2х100 мл) и 1 н. соляной кислотой (2х50 мл), растворитель отгоняют практически нацело (его вводят в рецикл без дополнительной очистки), остаток - вязкую стекловидную массу, обрабатывают 25 мл спирта (pH спиртовой части должен быть менее 4, в случае необходимости - добавляют кислоту), образовавшуюся суспензию охлаждают до 0-5oC, выдерживают при этой температуре 2 ч, отфильтровывают, осадок на фильтре промывают охлажденным спиртом и сушат при 50oC. Маточный и промывной спирт после перегонки используют в обороте.
б. При работе в растворителях, смешивающихся с водой (ацетон, ДМФА и т. п.), растворитель отгоняют, остаток растворяют в метаноле (этаноле), раствор обрабатывают активированным углем, уголь отфильтровывают, фильтрат подкисляют разбавленной соляной кислотой до pH 3-4, выпавший продукт отфильтровывают, промывают водой и спиртом и сушат до постоянного веса при 50oC.
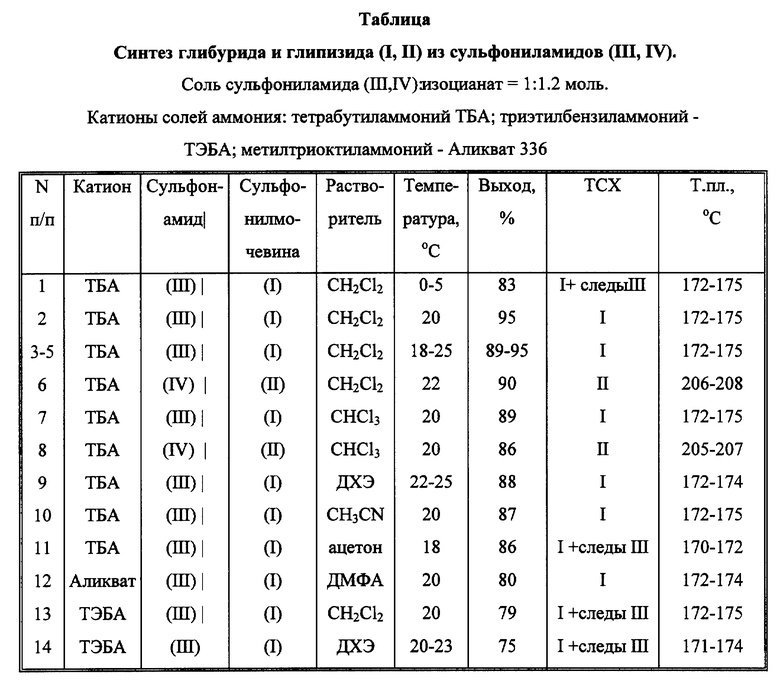
Аналогично осуществляли синтез и обработку реакции при варьировании параметров и реагентов; результаты приведены в таблице.
Состав и структура полученных соединений подтверждена их аналитическими и спектральными данными и совпадением температуры плавления полученных образцов с литературными данными.
Опыты 1, 2 характеризуют температурный режим; серия опытов 3-5 - статистический разброс выхода при масштабировании синтеза; оп. 6, 8 - синтез глипизида; оп. 7-11 - варьирование растворителя оп. 12-14 - влияние смены органического катиона.
В условиях оп. 2 синтезированы аналоги глибурида: 1-{4-[2-(2-метокси-5-хлорбензамидо)этил] фенилсульфонил} -3-(циклопентен- 2-ил)мочевина т.пл. 188-189oC (из метанола), выход 87%; 1-{4-[2-(2-метокси-5-хлорбензамидо)этил] фенилсульфонил} -3-(циклогептен- 2-ил)мочевина, т.пл. 142-144oC (из метанола), выход 90%; 1-{4-[2-(2-метокси-5-хлорбензамидо)этил]фенилсульфонил}-3-(4- метилциклогексил)мочевина т.пл. 189-190oC (из метанола), выход 89%; 1-{ 4-[2-(2-метокси-5-хлорбензамидо)этил]фенилсульфонил}-3- циклогептилмочевина, т.пл. 164-166oC (из метанола), выход 85%.
Источники информации
1. Машковский М. Д. Лекарственные средства. Изд. 13. Харьков: Торсинг, 1998. Т. 2. С. 23.
2. Черных В. П.//Хим.-Фарм.Ж. 1978. Т. 12. N 8. С. 13-20. Gerich J.E., Engl N. //J. Med. 1989. Vol. 321. P. 1231-1245. Липсон В.В., Полторак В.В., Горбенко Н.И.//Хим.-Фарм.Ж. 1997. Т. 31. N 11. С. 5-9.
3. Almuer W. et al. //Arzneim. Forsch. 1966. Bd 16. N 12. S. 1640-1642. Ambrogi V. et al.//Ibid. 1971. Db 21. N 2. S. 200-204.
4. Challinor-Rogers J/L/ et al.//J.Pharmacol. Exp. Ther. 1995. Vol. 273. N 2. P. 778-786.
5. Neth. Appl. 6.610.580 (1965).//C.A 1968. Vol. 68. 12733h. - Прототип.
6. Ger. (East) DD. 204,915. 1983.
7. Can. 889,876. 1972; Rom. 67.851. 1980, 67,848. 1983; Ger. (East) DD. 248,115. 1987.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения метсульфурон-метила | 2024 |
|
RU2824626C1 |
| Способ получения пиразосульфурон-этила | 2024 |
|
RU2828085C1 |
| Способ получения сульфонилмочевинных гербицидов, содержащих 4,6-диметоксипиримидин-2-ильный заместитель | 2024 |
|
RU2834461C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНИЛМОЧЕВИН ИЛИ ИХ СОЛЕЙ | 1973 |
|
SU399122A1 |
| Способ получения моносульфурон-метила | 2024 |
|
RU2834839C1 |
| Способ получения моносульфурона | 2024 |
|
RU2834000C1 |
| Способ получения R-N-[[3-[(диметиламино)карбонил]пиридин-2-ил]сульфонил]карбаматов, в которых заместителем R является метил или этил | 2023 |
|
RU2816572C1 |
| Способ получения никосульфурона | 2023 |
|
RU2807708C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛОВ АЛКОКСИФЕНИЛУКСУСНЫХ КИСЛОТ (ВАРИАНТЫ) И ГАЛОИДМЕТИЛЬНЫХ ПРОИЗВОДНЫХ ЭФИРОВ ФЕНОЛОВ | 2004 |
|
RU2273631C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИЗОХИНОЛИНА | 1970 |
|
SU417942A3 |
Описывается способ получения арилсульфонилмочевины, в том числе глибурида-1-{ 4-[2(2-метокси-5-хлорбензамидо)этил] фенилсульфонил}-3-циклогексилмочевины, и глипизида-1-{4-[2-(5-метилпиразин-2-карбоксамидо)этил]фенилсульфонил} -3-циклогексилмочевины, при взаимодействии соответствующих арилсульфонамидов с изоцианатами с последующим выделением целевых продуктов обычными способами, отличающийся тем, что арилсульфонамид вводят в реакцию в виде его органической соли в апротонном органическом растворителе, в том числе в растворителе, не смешивающемся с водой. Технический результат - упрощение процесса, увеличение чистоты и выхода целевого продукта. 3 з.п.ф-лы, 1 табл.
| Способ получения замещенной бензолсульфонилмочевины | 1970 |
|
SU464108A3 |
| Способ получения замещенной бензолсульфонилмочевины | 1970 |
|
SU488403A3 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОЛСУЛЬФОМОЧЕВИНЫ12 | 0 |
|
SU293340A1 |
| Способ получения полиарилентиохинодиимидов | 1983 |
|
SU1199765A1 |
| DE 1251753 A, 1967 | |||
| DE 3833439 A1, 1991 | |||
| US 5463081 A1, 1995 | |||
| Автоматические конвейерные весы | 1960 |
|
SU149592A1 |
