Изобретение относится к технологии тонкого органического синтеза и может быть использовано для получения 2,3,5,6- тетраалканоилоксигидрохинонов - дискоти- ческих жидких кристаллов.
Известен способ получения триметил- гидрохинона каталитическим гидрированием триметил-п-бензохинона. В качестве катализатора служит палладий, которые наносят на активированный уголь, сульфат или карбонат бария. В качестве растворителя используют алифатические кетоиы (ацетон, метилизобутилкетон, метилизопропилке- тон).
Таким образом, для получения производного гидрохинона из триметил-п-бензохинона используют дорогостоящий катализатор (палладий) и ведут синтез в опасных условиях (водород, давление).
Известен способ получения тетраза- мещенного гидрохинона реакцией восстановления тетразамещенного п-хинона водородом, который вводится в реакционную массу под давлением 500 - 5000 psig. Реакция протекает наиболее эффективно в ацетонитриле при 100 - 200°С в течение 15ч. Общий выход достигает -71 % (реакция восстановления проводится после реакции
V
00 СП
ч
VI
окисления, без выделения продукта). Таким образом, в смеси присутствуют побочные продукты (исходные и промежуточные). Следовательно, получение чистого конечного продукта с большим выходом по этому способу нереально. Высокая температура способствует увеличению в реакционной массе продуктов осмоления, применение водорода и высокого давления усложняет технологию получения тетразамещенного гидрохинона и опасно для обслуживающего персонала.
Известен также способ ацилироеания тетрагидроксихинона в избытке хлорангид- ридов жирных кислот нагреванием суспензии в масляной бане до температуры кипения соответствующего хлорангидрида и выдержкой при этой температуре в течение 2 ч, охлаждением и дальнейшим разбавлением в холодном содовом растворе, длительном перемешивании полученной эмульсии до затвердевания масла, фильтрацией осадка, промывкой его холодной водой и спиртом, перекристаллизацией из разбавленного или 95%-го спирта в зависимости от номера гомолога.
Полученные эфиры имеют ярко-желтую окраску, плавятся с разложением. Выход не указан. Длительность синтеза неопределенна, так как не известно, какой период времени надо нагревать до температуры кипения хлорангидрида и сколько времени перемешивать до затвердевания эфи- ров.
Методика опробована только для первых членов гомологического ряда, для более высоких номеров гомологов (п 5) она не выполнялась. Кроме того, по этому способу получается ацилированный тетрагидрокси- хинон и необходима дополнительная стадия по его восстановлению в гидрохинон. К недостаткам этого способа следует отнести также неконтролируемость начала реакции ацилирования и отсутствие операций по улавливанию выделяющегося хлористого водорода.
Наиболее близким по технической сущности и достигаемым результатам является способ получения тетразамещенного гидрохинона путем ацилирования тетрагидрокси- п-хинона хлорангидридом каприловой кислоты до тетраоктаноилокси-п-хинона-IV в среде пиридина при в течение 3 ч с последующим восстановлением продукта ацилирования раствором 2 н. HCI с получением восстановленной формы 2,3,5,6-тетра- октаноилоксихинон с выходом по первой стадии 20-30%, по второй 90-95% и общим выходом 18-23,5%, Продукт выделяют и очищают перекристаллизацией из этанола
хроматографической очисткой из смеси гек- сана с ацетоном и последовательной перекристаллизацией из гексана с ацетоном.
Недостатками данного способа являют- ся небольшой общий выход конечного продукта, двустадийность методики, относительно большое время синтеза, неконтролируемость кислотности среды.
Цель изобретения - повышение выхода 0 целевого продукта и интенсификация процесса.
Поставленная цель достигается путем взаимодействия 2,3,5,6-тетрагидроксихино- на с хлорангидридом алифатической карбо- 5 новой кислоты формулы л
CHyfcHJnCCct где , в среде смешанного растворителя
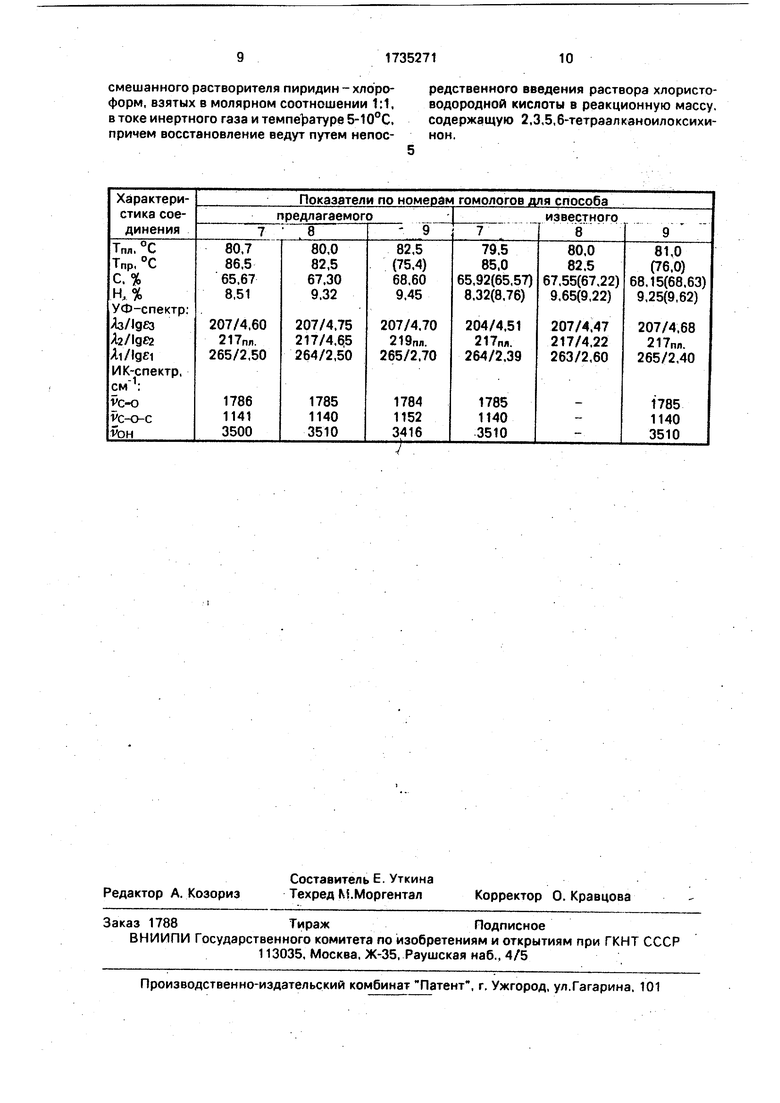
пиридин-хлороформа 1:1 в токе инертного газа при температуре 5-10°С с последую0 щим восстановлением промежуточного 2,3,5,6-тетраалканоилоксихинона 2N водным раствором HCI, при этом кислота непосредственно вводится в реакционную массу, содержащую хинон. Продукт выделя5 ют перекристаллизацией из этанола, хроматографической очисткой из смеси гексана и ацетона и последовательной перекристаллизацией из этанола и гексана. Выход после очистки 45-46% от теоретического.
0 Отличительными признаками процесса являются проведение взаимодействия тетрагидроксихинона с хлорангидридом кар- боновой кислоты в среде смешанного растворителя пиридин - хлороформ. взятых
5 в молярном соотношении 1:1 в токе инертного газа и температуре 5-10°С и проведение восстановления путем непосредственного введения раствора хлористоводородной кислоты в реакционную массу, со0 держащую хинон, что позволяет повысить выход целевого продукта и интенсифицировать процесс.
Пример 1. В пятигорлую колбу объемом 0,5 л, снабженную мешалкой, термо5 метром, обратным холодильником, капельной воронкой, капилляром для введения инертного газа и хлоркальциевой трубкой, вводят 5,2 г (0,03 моль, ММП72М) тетрагидроксихинона, растворенного в 0,5
0 моль сухого пиридина (d Oi98, ,11), и добавляют 0,5 моль хлороформа (,37;d 1,50). В реакционную массу, охлаждая ее, в течение 30 мин пропускают инертный газ. Температура снижается до
5 -5°С. Затем в течение 15 мин закапывают 19,5 г (0,12 моль, ,66) хлорангидрида каприловой кислоты, поддерживая температуру 5°С, выдерживают реакционную массу при данной температуре при перемешивании еще 30 мин. Затем без охлаждения
в течение 30 мин закапывают 2 н, растбор хлористоводородной кислоты в количестве 160 мл до момента расслоения хлороформного и водного слоев. Температура повышается до 25-35°С, цвет реакционной массы изменяется от желто-оранжевого до темно- вишневого; Прекращают подачу газа, хлороформный слой отделяют от водного. Обрабатывают водный слой дополнительно двумя порциями хлороформа по 25 мл, хло- реформ отгоняют. Пастообразную массу растворяют при нагревании в четырехкратном количестве этанола. После охлаждения выделяют мелкие кристаллы 2,3,5,6-тетра- октаноилоксигидрохинона, которые,допол- нительно подвергают хроматографической очистке из смеси гексана с ацетоном 95:5 на силикагеле, перекриста/1лизовываютиэ этанола, затем из гексана. Получают 9,05 г 2,3,5,6-тетраоктаноилоксигидрохинона, вы- ход 40%.
Пример 2. В пятигорлую колбу объемом 0,5 л, снабженную мешалкой, термометром, обратным холодильником, капельной воронкой, капилляром для введения инертного газа и хлоркальциевой трубкой, вводят 5,2 г (0,03 моль, ,10) тетрагидроксихинона, растворенного в 0,5 моль сухого пиридина (d 0,98; ММ 79,11), и добавляют 0,5 моль хлороформа ,37; ,50). В реакционную массу, охлаждая ее, в течение 30 мин пропускают инертный газ. Температура снижается до -5°С. Затем в течение 15 мин закапывают 19,5 г (0,12 моль; ,66) хлорангидри- да каприловой кислоты, поддерживая температуру 10°С, выдерживают при данной температуре реакционную массу при перемешивании еще 30 мин. Затем без охлажде- ния в течение 30 мин закапывают 2 н. раствор хлористоводородной кислоты в количестве 160 мл до момента расслоения хлороформного и водного слоев. Температура повышается до 25-30°С, цвет реакционной массы изменяется от жел го-оранжевого до темно-вишневого. Прекращают подачу газа, хлороформный слой отделяют от водного, Обрабатывают водный слой дополнительно двумя порциями хлорофор- ма по 25 мл, хлороформ отгоняют. Пастообразную массу растворяют при нагревании в четырехкратном количестве этанола. После охлаждения выделяют кристаллы 2,3,1,6- тетраалканоилоксигидрохинона, которые дополнительно подвергают хроматографической очистке из смеси гексана с ацетоном 95:5 на силикагеле, перекристаллизовыва- ют из этанола, затем из гексана. Получают 8,6 г продукта. Выход 38%.
Пример 3. В пятигорлую колбу, снабженную мешалкой, термометром, обратным холодильником, капельной воронкой, капилляром для введения инертного газа и хлоркальциевой трубкой, вводят 5,2 г (0,03 моль, ,10) тетрагидроксихинона, растворенного в 0,5 моль сухого пиридина (d - 0,98; ,11), и добавляют 0,5 моль хлороформа (d 1,5; ,37). В реакционную массу, охлаждая ее, в течение 30 мин пропускают инертный газ. Температура снижается до -5°С. Затем закьпывают 19,5 г (,66, 0,12 моль) хлорангидрида каприловой кислоты в тече- , ние 15 мин поддерживая температуру 8°С и, перемешивая при данной температуре, выдерживают реакционную массу еще 30 мин. Затем без охлаждения в течение 30 мин закапывают 2 н. раствор хлористоводородной кислоты в количестве 160 мл до момента расслоения хлороформного и водного слоев. Температура повышается до 25-30°С, цвет реакционной массы изменяется от желто-оранжевого до темно-вишневого. Прекращают подачу газа, хлороформный слой отделяют от водного. Обрабатывают водный слой еще дважды хлороформом по 25 мл, хлороформ отгоняют. Пастообразную массу растворяют при нагревании в четырехкратном количестве этанола. После охлаждения выделяются мелкие кристаллы 2,3,5,6-тетраоктаноилоксигидрохинона, которые дополнительно подвергают хроматографической очистке из смеси гексана с ацетоном 95:5 на силикагеле, перекристал- лизовывают из этанола, затем из гексана. Получают 9,7 г 2.3,5,6-тетраоктаноилокси- гидрохинона. Выход от теоретического 43%.
Пример 4. В пятигорлую 0,5 л колбу, снабженную мешалкой, термометром, обратным холодильником, капельной воронкой, капилляром для введения инертного газа и хлоркальциевой трубкой, вводят 5,2 г (0,03 моль, ,10) тетрагидроксихинона, растворенного в 0,5 моль сухого пиридина (d 0,98; ММ 79,11), и добавляют 0.5 моль хлороформа (ММ 119,37; d 1,50), В реакционную массу, охлаждая ее в течение 30 мин, пропускают инертный газ. Температура снижается до -5°С. Затем закапывают 19.5 г (,66; 0,12 моль) хлорангидрида каприловой кислоты в течение 15 мин при температуре 15°С. Затем при этой температуре выдерживают реакционную массу еще 30 мин, перемешивая. Далее без охлаждения закапывают 2н. раствор хлористоводородной кислоты в количестве 160 мл в течение 30 мин до момента расслоения хлороформного и водного слоев. Температура повышается до 25-30°С, цвет реакционной массы изменяется от желто-оранжевого до темно-вишневого. Прекращают подачу газа, хлороформный слой отделяют от водного. Обрабатывают водный слой еще дважды хлороформом по 25 мл, хлороформ отгоняют. Пастообразную массу растворяют при нагревании в четырехкратном количестве этанола. После охлаждения выделяются мелкие кристаллы 2,3,5,6-тет- раоктаноилоксигидрохинона, которые дополнительно подвергают хроматографи- ческой очистке из гексана с ацетоном - 95:5, перекристаллизовывают из этанола, а затем изгексана. Получают 8,15 г продукта. Выход 36%.
Пример 5. В пятигорлую 0,5 л колбу, снабженную мешалкой, термометром, обратным холодильником, капельной воронкой, капилляром для введения инертного газа и хлоркальциевой трубкой, вводят 5,2 г (0,03 моль, ,10) тетрагидроксихино- на, растворенного в 0,5 моль сухого пиридина (,98; ,11), и добавляют 0,5 моль хлороформа (,37; ,50). В реакционную массу, охлаждая ее до -5°С, пропускают инертный газ в течение 30 мин. Затем закапывают 17,22 г (MMH48J; 0.12 моль) хлорангидрида энантовой кислоты в течение 15 мин при температуре 10°С, затем, перемешивая, при этой температуре выдерживают реакционную смесь еще 30 мин. Затем без охлаждения закапывают 2н. раствор хлористоводородной кислоты в количестве 160 мл в течение 30 мин до момента расслоения хлороформного и водного слоев. Температура повышается до 25-30°С, цвет реакционной массы изменяется от желто- оранжевого до темно-вишневого, прекращают подачу газа, хлороформный слой отделяют от водного, еще дважды водный слой обрабатывают хлороформом по 25 мл, хлороформ отгоняют. Пастообразную массу растворяют при нагревании в четырехкратном количестве этанола. После охлаждения выделяются кристаллы 2,3,5,6-тетрагепта- ноилоксигидрохинона, которые дополнительно подвергают хроматографической очистке из гексана с ацетоном - 95:5, перекристаллизовывают из этанола, а затем из гексана. Получают 9,13 г продукта. Выход 45%.
Пример 6. В пятигорлую 0,5 л колбу, снабженную мешалкой, термометром, обратным холодильником, капельной воронкой, капилляром для введения инертного газа и хлоркальциевой трубкой, вводят 5,2 г (0,03 моль; ,10) тетрагидроксихино- на, растворенного в 0,5 моль сухого пиридина (,98: ,11), и добавляют 0,5 моль
хлороформа (,37; ,5), В реакционную массу, охлаждая ее, в течение 30 мин пропускают инертный газ. Затем закапывают 21,21 г (,81; 0,12 моль) хлорангидрида пеларгоновой кислоты в течение 15 мин при температуре 80°С. Перемешивая при этой температуре, выдерживают реакционную массу еще 30 мин. Затем без охлаждения закапывают 2н, раствор
хлористоводородной кислоты в количестве 160 мл в течение 30 мин до момента расслоения .хлороформного и водного слоев. Температура повышается до 30°С, цвет меняется от желто-оранжевого до вишневого, прекращают подачу газа, хлороформный слой отделяют от водного, обрабатывают водный слой еще дважды хлороформом по 25 мл, хлороформ отгоняют, Пастообразную массу растворяют при
нагревании в четырехкратном количестве этанола. После охлаждения выделяются кристаллы 2,3,5,6-тетранонэноилокси- гидрохинона, которые затем хроматогра- фируют из гексана с ацетоном - 95:5,
перекристаллизовывают из этанола, затем из гексана. Получают 9,6 г целевого продукта. Выход 40%.
Формула изобретения Способ получения жидкокристаллических гомологов 2,3,5.6-тетраалканоилокси- гидрохинонов общей формулы
О
1
.О-СЧВД
где
iffi-0
п 5-7, (J
о-иадл
путем взаимодействия 2,3,5,6-тетрагидрок- сихинона с хлорангидридом алифатической карбоновой кислоты формулы
О
ШИЭДа-С
где п 5-7,
в присутствии пиридина с последующим восстановлением промежуточного 2,3,5,6- тетраалканоилоксихинона 2N водным рас0 твором хлористоводородной кислоты и последующим выделением и очисткой целевого продукта перекристаллизацией из этанола, хроматографической очисткой из смеси гексана с ацетоном и последователь5 ной перекристаллизацией из этанола и гексана, отличающийся тем. что, с целью повышения выхода целевого продукта и интенсификации процесса, взаимодействие тетрагидроксихинона с хлорангидридом карбоновой кислоты проводят в среде
смешанного растворителя пиридин - хлороформ, взятых в молярном соотношении 1:1, в токе инертного газа и температуре 5-10°С, причем восстановление ведут путем непосредственного введения раствора хлористоводородной кислоты в реакционную массу, содержащую 2,3,5,6-тетраалкзноилоксихи- нок.



| название | год | авторы | номер документа |
|---|---|---|---|
| 3-ФЕНОКСИФЕНИЛСОДЕРЖАЩИЕ 1,3-ДИКЕТОНЫ В КАЧЕСТВЕ ИСХОДНЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ ИХ ХЕЛАТНЫХ КОМПЛЕКСОВ С ИОНАМИ МЕДИ (II) И СПОСОБ ПОЛУЧЕНИЯ 3-ФЕНОКСИФЕНИЛСОДЕРЖАЩИХ 1,3-ДИКЕТОНОВ | 2012 |
|
RU2475473C1 |
| Способ получения производных 3-азабицикло 3,3,1 нонана,или их изомеров,или их фармацевтически приемлемых солей присоединения кислот (его варианты) | 1984 |
|
SU1395141A3 |
| Способ получения производных 8( -аминоэтил) эрголина-1 или их солей | 1975 |
|
SU565914A1 |
| Способ получения оптически активных производных оксазафосфорина | 1979 |
|
SU867314A3 |
| Способ получения производных 2-оксиметилз-3-окси-6-/1-окси2 аминоэтилпиридина или их солей | 1972 |
|
SU519130A3 |
| СПОСОБ ПОЛУЧЕНИЯ ВЕТА-КАРОТИНА | 1993 |
|
RU2074177C1 |
| Способ получения спиросоединения азолона или его N-оксидного производного или его основной соли с фармакологически применимым катионом | 1990 |
|
SU1838313A3 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАЛОИДОВ | 1972 |
|
SU351369A1 |
| Способ получения морфиновых производных или их солей | 1974 |
|
SU635868A3 |
| Способ получения кислородсодержащих гетероциклических соединений или их металлических солей | 1975 |
|
SU577999A3 |
Изобретение касается защищенных гидрохинонов, в частности 2,3,5,6-тетраал- кэноилоксигидрохинонов, где алканоил - группа: СНзЧСНаЬ- - С(0)-0-, используемых в качестве дискатических жидких кристаллов. Цель -увеличение выхода целевого продукта и интенсификация процесса. Его ведут ацилированием тетрагидрокси-п- - хинона хлорангидридом Су-Сэ-карбоно- вой кислоты в среде смеси пиридина и хлороформа (молярное соотношение 1:1) в токе инертного газа при 5-10°С с последующими восстановлением (без его выделения) 2 н. водным раствором HCI, выделением целевого продукта, перекристаллизацией из этанола, хроматографической пипеткой из смеси гексана и ацетона (95:5) и дальнейшей последовательной перекристаллизацией из этанола и гексана. Выход целевых продуктов в данном случае увеличивается в 1,5-2.5 раза (до 40-45%) при снижении времени синтеза в 2 раза. 1 табл. Ч-.Ё
